Terezinha Marta P.P. Castiñeiras, Luciana G. F. Pedro & Fernando S. V. Martins
Casos confirmados por local de ocorrência
Manifestações
A doença meningocócica é uma infecção bacteriana aguda, rapidamente fatal, causada pela Neisseria meningitidis. Esta bactéria pode causar inflamação nas membranas que revestem o sistema nervoso central (meningite) e infecção generalizada (meningococcemia). Existem 13 sorogrupos identificados de N. meningitidis, porém os que mais freqüentemente causam doença são o A, o B, o C, o Y e o W135.
Estima-se a ocorrência de pelo menos 500 mil casos de doença meningocócica por ano no mundo, com cerca de 50 mil óbitos. É uma doença de evolução rápida e com alta letalidade, que varia de 7 até 70%. Mesmo em países com assistência médica adequada, a meningococcemia pode ter uma letalidade de até 40%. Geralmente acomete crianças e adultos jovens, mas em situações epidêmicas, a doença pode atingir pessoas de todas as faixas etárias.
Transmissão
O ser humano é o único hospedeiro natural da N. meningitidis. Cerca de 10% dos adolescentes e adultos são portadores assintomáticos da bactéria na orofaringe ("garganta") e podem transmitir a bactéria, mesmo sem adoecer. A bactéria é transmitida de uma pessoa para outra pela secreção respiratória (gotículas de saliva, espirro, tosse). Geralmente, após a transmissão, a bactéria permanece na orofaringe do indivíduo receptor por curto período e acaba sendo eliminada pelos próprios mecanismos de defesa do organismo. Desta forma, a condição de portador assintomático tende a ser transitória, embora possa se estender por períodos prolongados de meses a até mais de um ano.
Em menos de 1% dos indivíduos infectados, contudo, a bactéria consegue penetrar na mucosa respiratória e atinge a corrente sanguínea levando ao aparecimento da doença meningocócica. A invasão geralmente ocorre nos primeiros cinco dias após o contágio. Os fatores que determinam o aparecimento de doença invasiva ainda não são totalmente esclarecidos.
Distribuição geográfica e riscos A doença meningocócica tem distribuição global, podendo ocorrer surtos ocasionais e epidemias em qualquer país do mundo. A África é a região com maior número de casos no mundo, principalmente na região semi-árida sub-Saariana, conhecida como “cinturão da meningite” que se estende do Senegal até a Etiópia, afetando cerca de 15 países. Nesta região, a doença meningocócica representa uma ameaça há, pelo menos, 100 anos com epidemias recorrentes a cada 8 a 12 anos, freqüentemente resultando em uma taxa de ataque 500 a 1000 vezes maior do que a de uma população em país desenvolvido.
As alterações climáticas influenciam a dinâmica da transmissão da doença meningocócica e as epidemias são mais freqüentes durante o inverno nas regiões temperadas e nas estações secas em regiões tropicais. Além disso, entre os sorogrupos a capacidade potencial de provocar epidemias é diferenciada. Os sorogrupos A e C têm a maior taxa de ataque, podendo chegar até 500 casos em cada 100 000 habitantes. Historicamente, o sorogrupo A foi o responsável pelas maiores epidemias e ainda atualmente provoca epidemias recorrentes no “cinturão da meningite” principalmente de novembro a junho, com redução do número de casos com o início da estação chuvosa. O sorogrupo B ocorre de forma endêmica em todos os continentes, inclusive nos países desenvolvidos, porém a taxa de ataque, durante uma epidemia, não ultrapassa 100 casos por 100000 habitantes.
A doença meningocócica pode ocorrer em pessoas de qualquer faixa etária, porém é mais comum em crianças até cinco anos e mais rara em idosos. Em geral, a incidência da doença é maior em países em desenvolvimento, especialmente em áreas com grandes aglomerados populacionais. A história de infecção recente pelo vírus influenza (gripe) e o tabagismo aumentam a chance de infecção meningocócica. Além disso, algumas pessoas por condições de doenças de base têm um maior risco de desenvolver a doença, como as submetidas à retirada cirúrgica do baço (esplenectomizados), ou as portadoras de disfunção do baço (asplenia funcional da anemia falciforme, da talassemia), ou aquelas com deficiências de imunoglobulinas e do complemento.
Cerca de 90% dos casos de doença meningocócica relatados no mundo são causados pelos sorogrupos A, B e C. Os sorogrupos B e C são responsáveis pela maioria dos casos na Europa e no Continente Americano e os sorogrupos A e C predominam na Ásia e África. A incidência do sorogrupo Y tem apresentado aumento significativo em alguns países, como em Israel, Suécia e Estados Unidos. O sorogrupo W-135 era uma causa rara de doença meningocócica, até que em 2000 foi descrito o primeiro surto causado por esse sorogrupo em peregrinos para Meca durante o Hajj (peregrinação islâmica anual). Nesse período, foram diagnosticados 241 casos de doença meningocócica (sorogrupo W135) na Arábia Saudita e 90 casos em viajantes (e em contactantes) após o retorno a 16 diferentes países de origem (Reino Unido, Bélgica, Estados Unidos, França, Marrocos, Kuait, Arábia Saudita, Oman, Indonésia, Singapura, Dinamarca, Finlândia, Suécia, Noruega, Alemanha e Holanda). O Hajj atrai mais de dois milhões de mulçumanos do mundo todo, formando um grande aglomerado populacional o que facilita a disseminação de doenças transmitidas por via respiratória, como a doença meningocócica. Em 2002, o sorogrupo W-135 foi introduzido na África, afetando cerca de 13 mil pessoas em Burkina Faso, com 1500 mortes.
No Brasil, a doença é endêmica com casos esporádicos durante todo o ano, principalmente no inverno, com surtos e epidemias ocasionais. As maiores epidemias registradas no país ocorreram na década de 70 e foram determinadas pelos sorogrupos A e C. Ao longo da década de 80, o sorogrupo B passou a ser o mais freqüente, com epidemia em 1988. Nos últimos 20 anos foram notificados, no Brasil, cerca de 80 mil casos, a maioria causada pelo sorogrupo B. O sorogrupo C aparece como o segundo mais freqüente, tendo sido responsável por alguns surtos, inclusive motivando vacinação em massa de crianças e adultos, como ocorreu em 1995. Em tazão disto, passados quase dez anos, com o aumento da população susceptível (pessoas nunca vacinadas e as que perderam a imunidade conferida pela vacina), o sorogrupo C volta a ser uma preocupação particularmente nos grande aglomerados urbanos como o Rio de Janeiro.
Tabela 1 - Doença meningocócica no Brasil: 1996 – 2005 Casos confirmados por local de ocorrência
| Região | 1996 | 1997 | 1998 | 1999 | 2000 | 2001 | 2002 | 2003 | 2004 | 2005* | Total |
| Norte | 358 | 240 | 295 | 393 | 270 | 318 | 285 | 263 | 248 | 145 | 2.815 |
| Nordeste | 1.415 | 1.356 | 1.297 | 1.066 | 1.270 | 1.058 | 900 | 725 | 718 | 568 | 10.373 |
| Sudeste | 4.198 | 3.401 | 3.163 | 2.586 | 2.530 | 2.040 | 1.873 | 1.662 | 2.007 | 1.773 | 25.233 |
| Sul | 1.074 | 998 | 958 | 922 | 720 | 643 | 675 | 597 | 562 | 355 | 7.504 |
| Centro-Oeste | 276 | 330 | 348 | 268 | 229 | 169 | 139 | 128 | 131 | 140 | 2.158 |
| Total | 7.321 | 6.325 | 6.061 | 5.235 | 5.019 | 4.228 | 3.872 | 3.375 | 3.666 | 2.981 | 48.083 |
* dados sujeitos à revisão.
Fonte: Ministério da Saúde - Secretaria de Vigilância em Saúde, 2006.
Fonte: Ministério da Saúde - Secretaria de Vigilância em Saúde, 2006.
Os fatores relacionados ao risco de adoecer não estão totalmente esclarecidos, contudo o contato próximo (Tabela 2) com pessoas infectadas é um fator de risco importante para o aparecimento de casos secundários. Estima-se que o risco de adoecimento entre os contactantes próximos é maior que o existente na população em geral, chegando a ser até 1000 vezes maior em pessoas que dividem o domicílio com o doente, o que justifica a adoção de medidas preventivas específicas direcionadas a este grupo.
Tabela 2 - Definição de contactante próximo
|
O risco exato da doença para viajantes é desconhecido. A incidência por mês de estada em países hiperendêmicos é estimada em 0,4 por 1000000 viajantes, porém as aglomerações populacionais tendem a aumentar a probabilidade de transmissão e, nas peregrinações à Meca, esta proporção pode chegar a 2000 por 1000000.
Medidas de proteção individual
De todas as doenças infecciosas, a doença meningocócica é uma das que causa maior impacto na população, pelo seu potencial de acometer de forma rápida e fulminante pessoas previamente saudáveis, na sua maioria crianças, e pelo risco de desencadear epidemias. A falta de informação adequada associada ao sensacionalismo oportunista colaboram para aumentar o pânico da população e não contribuem para o controle efetivo da doença. Algumas medidas, adotadas com alguma freqüência por motivos não muito claros, como fechamento de escolas e emergências, desinfecção de ambulâncias, são tecnicamente inadequadas, pois a bactéria não sobrevive no ambiente. Além disto são totalmente ineficazes para evitar ou controlar epidemias de doença meningocócica e, claramente, causam transtornos e prejuízos inclusive ao próprio atendimento médico à população.
O risco de doença meningocócica é mais significativo apenas para pessoas que tiveram contato muito próximo com uma pessoa infectada (portadora assintomática ou doente). Quando se detecta um novo caso (doente), admite-se que entre seus contactantes próximos, possam existir um ou mais portadores assintomáticos e, eventualmente, um outro indivíduo susceptível, que à semelhança do doente já identificado, possa adoecer gravemente (“vítima potencial”). A prevenção imediata da ocorrência de novos casos é feita através do tratamento profilático com antibióticos (quimioprofilaxia) de todos os contactantes próximos do indivíduo doente (Tabela 2), visando a eliminação da bactéria da nasofaringe dos portadores.
A definição de contactantes próximos pode ser variável de um país para outro e a identificação desses indivíduos, em geral, não é tarefa fácil e depende de uma investigação epidemiológica adequada. Não é incomum que todos os conhecidos de um indivíduo com doença meningocócica se julguem contactantes próximos e desejem receber quimioprofilaxia. Porém, a utilização da quimioprofilaxia em massa além de não ter impacto no controle da doença, não é isenta de riscos, pois os antibióticos utilizados para a profilaxia podem, eventualmente, estar associados com efeitos colaterais ou induzir o aparecimento de cepas bacterianas resistentes.
A quimioprofilaxia, quando indicada, deve ser iniciada o mais precocemente possível, de preferência nas primeiras 24 horas, pois a chance de um indivíduo evoluir com doença invasiva é maior nos primeiros cinco dias após a infecção. A eficácia da quimioprofilaxia, quando feita adequadamente, é de 90 – 95%. Portanto, mesmo os contactantes que receberam a quimioprofilaxia podem vir a adoecer e devem estar alerta para o aparecimento dos primeiros sintomas, pois o retardo no início do tratamento implica em maior letalidade.
Mesmo durante epidemias ou surtos, a quimioprofilaxia é recomendada apenas para os contactantes próximos. Nessa situação, deve ser considerada a utilização da vacina como medida profilática. Cabe aos serviços de vigilância epidemiológica, a identificação precoce de surtos e epidemias e a definição da população alvo para a vacinação.
A maioria das vacinas disponíveis contra a doença meningocócica é constituída por antígenos polissacarídicos da cápsula da bactéria e confere proteção por tempo limitado (cerca de três anos) e exclusivamente para os sorogrupos contidos na vacina, com reduzida eficácia em crianças de baixa idade (particularmente abaixo de 2 anos). As mais freqüentemente empregadas são a vacina bivalente (A+C), a tetravalente (A+C+Y+W135) e, no caso de menores de 2 anos, a monovalente A. Para a meningite meningocócica B nenhuma vacina desenvolvida até então (inclusive a "cubana") mostrou-se eficaz de forma inequívoca. Mais recentemente foi desenvolvida uma vacina conjugada para a meningite meningocócica C, com elevada eficácia, proteção prolongada (possivelmente por toda a vida) e boa resposta em menores de um ano. Alguns países desenvolvidos, como a Inglaterra, já adotaram esta vacina de forma rotineira no calendário vacinal infantil.
No Brasil, as vacinas antimeningocócicas estão disponíveis na Rede Pública apenas em situações de surtos e epidemias. A vacina conjugada C está disponível nos Centros de Referência para Imunobiológicos Especiais (CRIE) exclusivamente para pessoas a partir dos 2 meses de idade e que tenham doenças ou condições de base que impliquem em um maior risco de doença meningocócica (asplenia congênita ou adquirida, esplenectomia, deficiências de complemento, anemia falciforme e talassemia). A solicitação deve ser feita pelo médico através de uma Ficha de Encaminhamento. Na rede privada, podem ser encontradas as vacinas bivalente A+C e a conjugada C.
Os viajantes que se dirigem para áreas hiperendêmicas de doença meningocócica, como o “cinturão da meningite na África”, devem ser vacinados, de preferência, com a vacina tetravalente (A+C+Y+W135) pelo menos 14 dias antes de viajar. Em 2002, o governo da Arábia Saudita passou a exigir a vacina antimeningocócica tetravalente, para concessão de vistos para os peregrinos que se dirigem à Meca durante o Hajj.
Manifestações
A doença meningocócica tem início abrupto e evolução rápida, podendo levar ao óbito em menos de 24 a 48 horas. As manifestações iniciais da meningite são febre alta, prostração, dor de cabeça, vômitos, aparecimento na pele de pequenas manchas violáceas (petéquias) que inicialmente são semelhantes às picadas de mosquitos mas que rapidamente aumentam de número e de tamanho, dor e dificuldade na movimentação do pescoço (rigidez de nuca). Em crianças com menos de um ano de idade, as manifestações da meningite podem ser mais inespecíficas como febre, irritação, choro constante e abaulamento da fontanela (“moleira”) sem rigidez de nuca. Se não for rapidamente tratada com antibióticos, a doença pode evoluir com confusão mental e coma. A meningococcemia é a forma mais grave de apresentação da infecção pela N. meningitidis e as manifestações iniciais são semelhantes às da meningite, excluindo-se a rigidez de nuca. O risco maior da doença meningocócica é a evolução rápida para o choque (diminuição acentuada da pressão arterial), o que resulta em funcionamento inadequado de órgãos vitais (como os rins, coração e pulmão) e morte. Cerca de 15 a 20% dos casos apresentam meningococcemia sem meningite, que tem letalidade próxima de 70% em países em desenvolvimento.
Doença meningocócica: petéquias
Foto: Luciana G. F. Pedro, 2004.
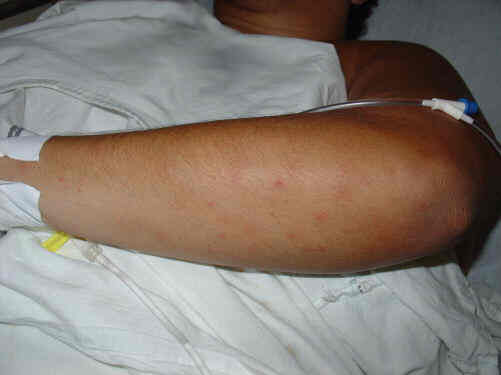 | Petéquias em antebraço esquerdo, semelhantes a picadas de mosquito |
A doença meningocócica pode ser confundida com outras doenças infecciosas como, por exemplo, o dengue grave (“hemorrágico”), embora a diferenciação seja simples. A pessoa com dengue pode ficar grave quando a febre começa a desaparecer, o que geralmente acontece depois do terceiro dia de doença, enquanto na doença meningocócica os sinais de gravidade aparecem em menos de 24 a 48 horas. Além disto existem outros agentes infecciosos que podem causar meningite (outras bactérias, vírus, fungos, etc). Nesses casos, as manifestações clínicas podem ser semelhantes e a diferenciação depende da realização de exames complementares. Quando existe a suspeita de doença meningocócica, o início do tratamento deve ser imediato e não é possível aguardar os resultados. A letalidade da doença meningocócica, se não tratada precocemente com antibióticos adequados, é virtualmente de 100%.
O diagnóstico inicial de doença meningocócica é clínico (história + exame físico da pessoa), feito essencialmente por exclusão de outras doenças. A confirmação laboratorial definitiva do diagnóstico é usualmente feita através do isolamento em cultivo da Neisseria meningitidis a partir de amostras de sangue ou de líquido céfalo-raquidiano (obtido por punção lombar), o que requer um certo tempo (1 a 3 dias). Contudo, a demonstração direta da presença da bactéria em amostras de líquor ou raspado de lesão cutânea através da coloração pela técnica do Gram (exame simples e rápido) permite aumentar o grau de certeza do diagnóstico clínico.
A doença meningocócica é uma emergência médica. Nos casos suspeitos, a pessoa deverá ser levada rapidamente para unidade de saúde mais próxima para avaliação médica. Mesmo que não haja recursos para confirmação diagnóstica, é de responsabilidade da unidade que prestou o primeiro atendimento o início imediato do tratamento com antibiótico adequado e hidratação. Após o início do tratamento o doente poderá ser transferido para um hospital de referência, obrigatoriamente em ambulância com acompanhamento médico. O contato telefônico prévio com o hospital de referência é imprescindível para se certificar da disponibilidade de vaga, evitando assim o deslocamento desnecessário e arriscado de uma pessoa em estado potencialmente crítico.
Tratamento Quando há suspeita clínica, o início do tratamento deve ser imediato e não deve aguardar resultados de exames. O tratamento é feito com antibióticos por via endovenosa e medidas de suporte (como hidratação). Cerca de 5 a 10% dos indivíduos evoluem para óbito apesar do tratamento. Das pessoas que sobrevivem, 9 a 11% ficam com algum tipo de seqüela permanente (surdez, paralisias, convulsões, amputação de extremidades). A administração de antibiótico profilático para os contactantes próximos dos indivíduos doentes deve ser feita rapidamente, pois reduz significativamente o aparecimento de casos secundários.


